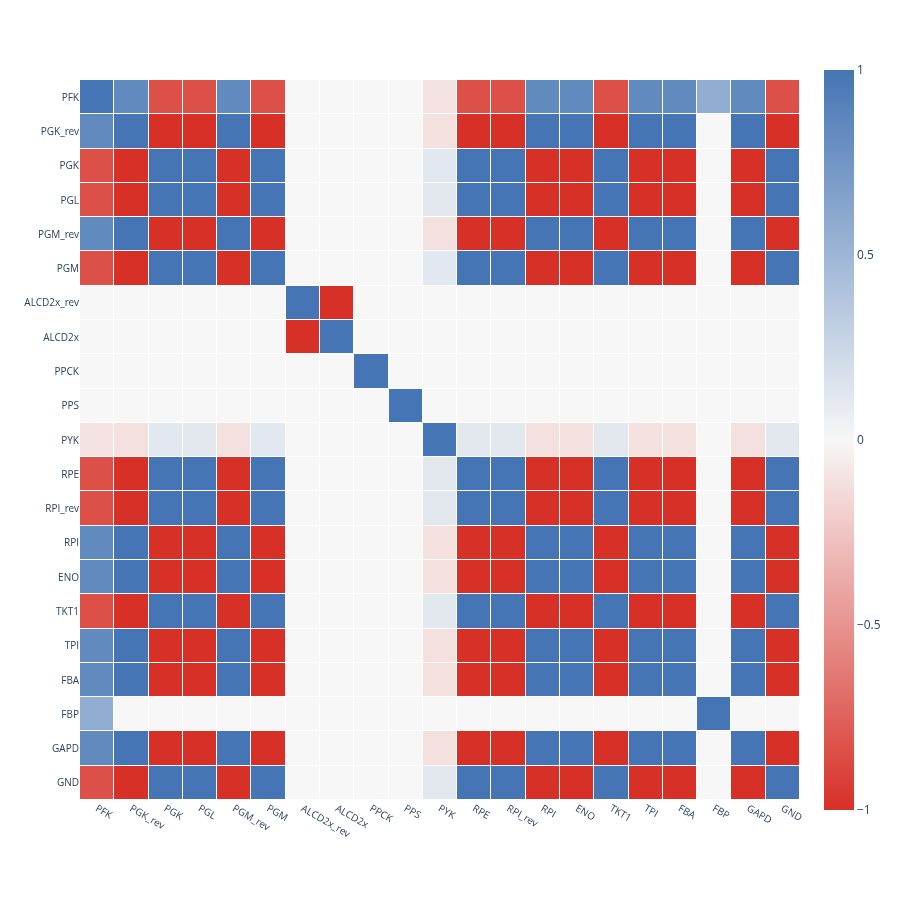
Correlations from reactions samples
Prerequisites for calculating pairwise correlations
In some cases, the forward direction of reactions (as defined in the model) does not align with the observed network topology. As a result, pairs of reactions may show identical correlation magnitudes but with opposite signs than the expected result.
To deal with this, we split all reversible reactions with both positive and negative flux to separate reactions. Thus, one of the 2 (forward or reverse) has the expected correlations with reactions from the rest network.
The split_forward_reverse function splits all bidirectional reactions having at least 1 positive and 1 negative flux value in a sampling array into separate forward and reverse reactions. This step is useful to avoid losing information, when computing correlations from a reactions set that includes reversible reactions.
steady_statesis the default flux Sampling array with only forward reactionsreactionsis a list with reactions of interest to be split and result in separate forward and reverse reactions. This list includes reactions from a pathway of interest and thus not all reactions from the list will be split (only those that have both positive and negative flux)
(subset_extended_steady_states_100,
subset_extended_reactions_100) = split_forward_reverse(
steady_states = subset_pathways_optgp_condition_100,
reactions = reactions_in_pathways_ordered)
(subset_extended_steady_states_0,
subset_extended_reactions_0) = split_forward_reverse(
steady_states = subset_pathways_optgp_condition_0,
reactions = reactions_in_pathways_ordered)
The find_reactants_products function identifies and saves in separate lists the reactants, products and directionality status of each reaction in a given metabolic model. Cofactors do not count as reactants/products.
cobra_modelis a metabolic model in cobra formatreactions_idsis a list of the reactions of interest
(reversibility_list_all_reactions_100,
reactants_list_all_reactions_100,
products_list_all_reactions_100) = find_reactants_products(
cobra_model = ec_cobra_model,
reactions_ids = subset_extended_reactions_100)
(reversibility_list_all_reactions_0,
reactants_list_all_reactions_0,
products_list_all_reactions_0) = find_reactants_products(
cobra_model = ec_cobra_model,
reactions_ids = subset_extended_reactions_0)
The sharing_metabolites_square_matrix function works given the lists with reactants, products and reversibility information, creates a square boolean matrix with True values representing reactions sharing a common metabolite, as implemented in sharing_metabolites helper function.
reactions_idsis a list containing IDs from a set of reactions of interestreversibility_list_all_reactionsis a list containing the reversibility status from a set of reactions (reactions must be the same and in accordance with their order inreactions_idslist. This is ensured with thesort_reactions_by_model_orderfunction)reactants_list_all_reactionsis a list containing the reactants from a set of reactions (reactions must be the same and in accordance with their order inreactions_idslist. This is ensured with thesort_reactions_by_model_orderfunction)products_list_all_reactionsis a list containing the products from a set of reactions (reactions must be the same and in accordance with their order inreactions_idslist. This is ensured with thesort_reactions_by_model_orderfunction)
subset_boolean_sharing_metabolites_matrix_100 = sharing_metabolites_square_matrix(
reactions_ids = subset_extended_reactions_100,
reversibility_list_all_reactions = reversibility_list_all_reactions_100,
reactants_list_all_reactions = reactants_list_all_reactions_100,
products_list_all_reactions = products_list_all_reactions_100)
subset_boolean_sharing_metabolites_matrix_0 = sharing_metabolites_square_matrix(
reactions_ids = subset_extended_reactions_0,
reversibility_list_all_reactions = reversibility_list_all_reactions_0,
reactants_list_all_reactions = reactants_list_all_reactions_0,
products_list_all_reactions = products_list_all_reactions_0)
Calculation of pairwise linear correlations and non-linear copula dependencies
The correlated_reactions function calculates pairwise linear correlations and non-linear copula dependencies given a flux sampling array. Users can choose the preffered coefficient to calculate pairwise correlations (pearson or spearman). Moreover, users can filter correlations not meeting a cutoff value and choose whether to proceed with calculation of non-linear dependencies using copulas. Correlations and dependencies from reactions not sharing metabolites can also be removed.
steady_statesis a numpy array of the generated steady states fluxesboolean_sharing_metabolites_matrixis a boolean symmetric numpy 2D array withTrue/Falsebased on the presense of shared metabolites between reactions. This matrix, which is optionally provided, is calculated with thesharing_metabolites_square_matrixfunctionreactionsis a list with the reactions IDs (must be in accordance with the rows of the steady states)linear_coeffis a string declaring the linear coefficient to be selected. Available options: “pearson”, “spearman”linear_corr_cutoffis a cutoff to filter (remove) linear correlations (not greater than the cutoff) based on the pearson or spearmanr coefficientindicator_cutoffis a cutoff to classify non-linear correlations as positive, negative or non-significantjensenshannon_cutoffis a cutoff to filter (remove) non-linear correlations (not greater than the cutoff) based on the Jensen-Shannon metricstd_cutoffis a cutoff to avoid computing the copula between 2 fluxes with almost fixed valuesinclude_non_linearis a boolean variable that ifTrue, takes into account and calculates non-linear copula dependenciescellsis the number of cells to compute the copulacop_coeffis a value that narrows or widens the width of the copula’s diagonal (use lower values to capture extreme tail dependences)lower_triangleis a boolean variable that ifTruereturns only the lower triangular matrix (useful for heatmap visualization)verboseis a boolean variable that ifTrueadditional information is printed as an output to the user.
If user requests calculation of non-linear copula dependencies, 3 numpy arrays and 1 dictionary are returned, as shown below.
(subset_linear_correlation_matrix_100,
subset_non_linear_correlation_matrix_100,
subset_mixed_correlation_matrix_100,
subset_correlations_dictionary_100) = correlated_reactions(
steady_states = subset_extended_steady_states_100,
boolean_sharing_metabolites_matrix = None,
reactions = subset_extended_reactions_100,
linear_coeff = "pearson",
linear_corr_cutoff = 0.3,
indicator_cutoff = 1.2,
jensenshannon_cutoff = 0.05,
std_cutoff = 1e-2,
include_non_linear = True,
cells = 5,
cop_coeff = 0.2,
lower_triangle = False,
verbose = True
)
(subset_linear_correlation_matrix_0,
subset_non_linear_correlation_matrix_0,
subset_mixed_correlation_matrix_0,
subset_correlations_dictionary_0) = correlated_reactions(
steady_states = subset_extended_steady_states_0,
boolean_sharing_metabolites_matrix = None,
reactions = subset_extended_reactions_0,
linear_coeff = "pearson",
linear_corr_cutoff = 0.3,
indicator_cutoff = 1.2,
jensenshannon_cutoff = 0.05,
std_cutoff = 1e-2,
include_non_linear = True,
cells = 5,
cop_coeff = 0.2,
lower_triangle = False,
verbose = True
)
If user does not request calculation of non-linear copula dependencies, 1 numpy array and 1 dictionary are returned, as shown below.
(subset_linear_correlation_matrix_100,
subset_correlations_dictionary_100) = correlated_reactions(
steady_states = subset_extended_steady_states_100,
boolean_sharing_metabolites_matrix = None,
reactions = subset_extended_reactions_100,
linear_coeff = "pearson",
linear_corr_cutoff = 0.3,
indicator_cutoff = 1.2,
jensenshannon_cutoff = 0.05,
std_cutoff = 1e-2,
include_non_linear = False,
cells = 5,
cop_coeff = 0.2,
lower_triangle = False,
verbose = True
)
(subset_linear_correlation_matrix_0,
subset_correlations_dictionary_0) = correlated_reactions(
steady_states = subset_extended_steady_states_0,
boolean_sharing_metabolites_matrix = None,
reactions = subset_extended_reactions_0,
linear_coeff = "pearson",
linear_corr_cutoff = 0.3,
indicator_cutoff = 1.2,
jensenshannon_cutoff = 0.05,
std_cutoff = 1e-2,
include_non_linear = False,
cells = 5,
cop_coeff = 0.2,
lower_triangle = False,
verbose = True
)
The dictionary returned from this function is independent and not filtered from the boolean_sharing_metabolites_matrix. It contains information on pairwise reactions linear coefficient values, the copula’s jensen shannon distance value, the copula’s indicator value and a copula classification value.
print(subset_correlations_dictionary_100.get("PYK~PGK"))
print(subset_correlations_dictionary_0.get("PYK~PGK"))
{'pearson': 0, 'jensenshannon': 0.11517866713970133, 'indicator': 1.6059379192742054, 'classification': 'positive_upper_lower_tail'}
{'pearson': 0, 'jensenshannon': -0.10089110481651221, 'indicator': 0.7628205134721049, 'classification': 'negative_upper_lower_tail'}
The plot_correlation_matrix function plots a correlation matrix created with the correlated_reactions function.
correlation_matrixis the correlation matrix to plot.reactionsis an optional list of reaction labels to be plot as axes in the plot.label_font_sizeis the font size for the axes labelswidthis the width of the plotheightis the height of the plot
plot_correlation_matrix(
correlation_matrix = subset_mixed_correlation_matrix_100,
reactions = subset_extended_reactions_100,
label_font_size = 10,
width = 900,
height = 900)